Ədəbiyyat siyahısının son yenilənmə tarixi: iyul 2016-ci il. Məqalənin son yenilənmə tarixi: mart 2015-ci il.
Məqalənin növbəti yenilənmə tarixi: noyabr 2017-cı il.
GİRİŞ.
Kardiomiopatiyalar ürək əzələlərinin xəstəlikləridir. Kardiomiopatiyalara miokardın müxtəlif xəstəlikləri daxildir ki, onların bir çoxu fərqli struktur və funksional fenotiplərlə müşahidə edilir və çox vaxtı genetikdir. Bir sıra mütəxəssis tərəfindən ürək-damar xəstəlikləri (məs., hipertoniya, ürəyin işemik xəstəliyi və ya ürəyik klapan xəstəliyi) səbəbindən yaranmış miokard xəstəliyini də kardiomiopatiyalar aid edir. Lakin, əksər aparıcı cəmiyyətlərin təriflərinə əsasən ürək-damar pozuntuları səbəbindən inkişaf etmiş ürək xəstəliyini kardiomiopatiyalara daxil etmir.
ƏSAS MEYARLAR.
1980-cı ildə Ümumdünya Səhiyyə təşkilatı tərəfindən kardiomiopatiyaların tərifi verilmişdir. Bu tərifə əsasən kardiomiopatiya "naməlum səbəblərdən ürək əzələsinin xəstəliyi" kimi müəyyən edilmişdir. Məlum ürək-damar xəstəlikləri səbəbindən (məs., hipertoniya, ürəyin işemik xəstəliyi və ya ürəyik klapan xəstəliyi) inkişaf edən ürək əzələsinin pozuntusu isə kardiomiopatiyadan fərqləndirilmişdir. Kliniki praktikada isə kardiomiopatiya termini həm də müxtəlif ürək-damar xəstəlikləri nəticəsində inkişaf etmiş miokardın xəstəliklərinə də aid edilir.
Bunun nəticəsində 1995-ci ildə ÜST və Kardiologiya üzrə beynəlxalq Cəmiyyət və Federasiyanın işçi qrupu tərəfindən kardiomiopatiyanın təsnifatı genişləşdirilmiş və ürək əzələsinə təsir göstərən bütün xəstəliklər buraya daxil edilmişdir. Üstəgəl təsnifatın aparılmasında həm kardiomiopatiyanın etiologiyası, həm də dominant patofiziologiya da nəzərə alınmışdır. 1995-ci ilin təsnifatına əsasən kardiomiopatiya "ürəyin disfunksiyası ilə müşaiyət olunan miokardın xəstəliyi" kimi təyin edilmişdir. Həmin təsnifata əsasən kardiomiopatiyalar aşağıdakı növlərə bölünmüşdür.
- Dilatasionı kardiomiopatiya (DKM)
- Hipertrofik kardiomiopatiya (HKM)
- Restriktiv kardiomiopatiya (RKM)
- Sağ mədəciyin aritmogen kardiomiopatiyası/displaziyası (SMAKM/D)
- Klassifikasiya olunmayan kardiomiopatiyalar
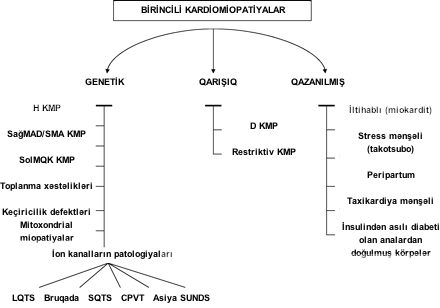
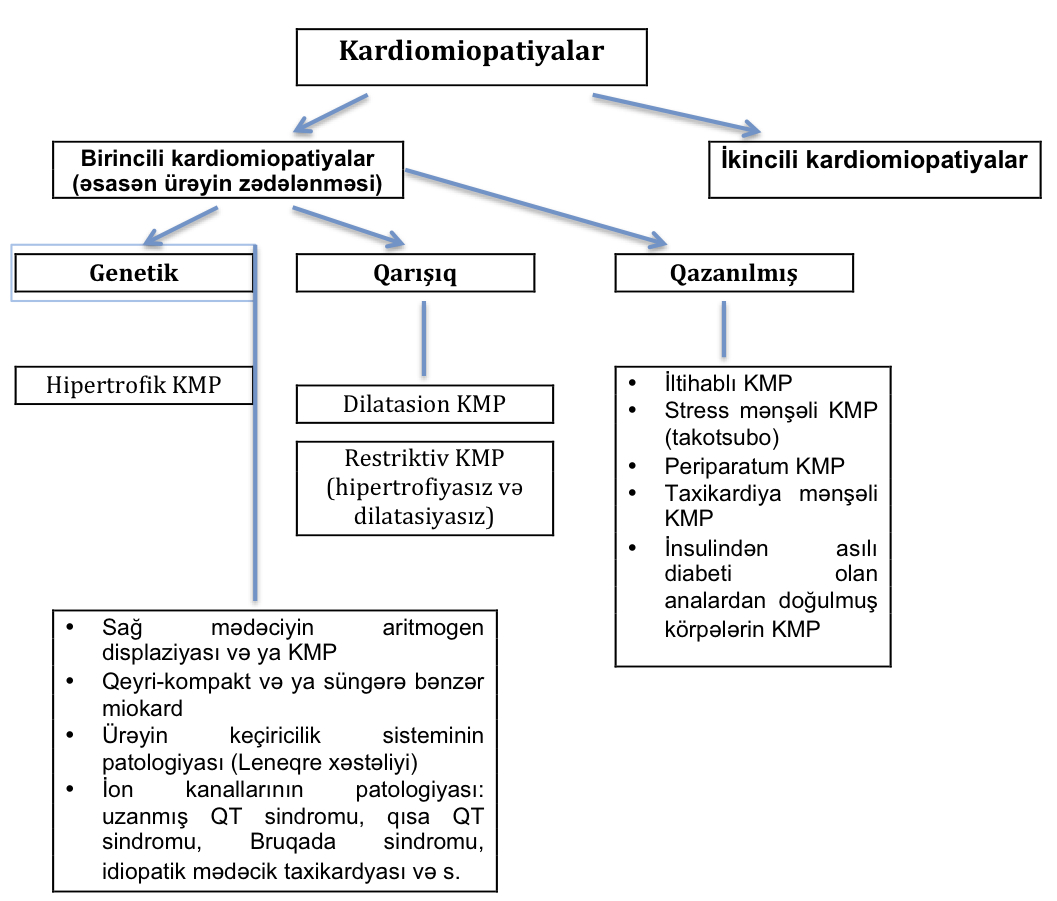
- birincili kardiomiopatiyalar (əsasən ürək pozuntusu ilə əlaqələndirilir) və
- ikincili kardiomiopatiya (digər orqanlar sistemində pozuntularla müşaiyət olunur).
- genetik: hipetrofik kardiomiopatiya, sağ mədəciyin aritmogen displaziyası (kardiomiopatiyası), sol mədəciyin süngərəbənzər kardiomiopatiyası (miokard əzələsi liflərinin anadangəlmə sıxlaşma qüsuru), PRKAG2 və Danon qlikogen depozit xəstəlikləri, ürəyin keçiricilik defektləri, mitoxondrial miopatiyalar və ion kanal pozuntuları.
- qarışıq (əsasən qazanılmış, az hallarda genetik): diltasion və restriktiv kardiomiopatiyalar.
- qazanılmış: miokardit, stress mənşəli (takotsubo) kardiomiopatiya, taxikardiya mənşəli və insulin asıllı diabeti olan analardan doğulmuş körpələrin kardiomiopatiyası.
- İnfiltrativ: amiloidoz, Quşer, Hurler və Hanter xəstəlikləri
- Toplanma xəstəlikləri: Fabri, Qlikogen toplanma xəstəliyi, Niman-Pik xəstəliyi, Hemoxromatoz
- Toksiklik: dərmanlar, ağır metallar
- Endomiokardial: endomiokardial fibroz, Loeffler endokarditi
- İltihablı: sarkoidoz
- Endokrin: diabet, hipertireoz, hipotireoz, hiperparatireoz, feoxromositoma, Akromeqaliya
- Kardiofasial: Noonan, Lentiginoz
- Neyro-əzələ: Fridrix ataksiyası, Dyuşen-Beker əzələ distrofiyası, miotonik distrofiya
- Metabolik-Qidalanma pozuntuları: Beri-beri, sinqa, selenium çatışmazlığı
- Autoimmun: SLE, dermatomiozit, skleroderma
- Xərçəngə qarşı terapiyanın fəsadları: antrasiklinlər, şüalanma, siklofosfamid
- Sol mədəciyin genişlənməsi: bu atım həcminin artması ilə nəticələnir.
- Frank-Starlinq münasibəti: mədəcik divarı dartıldıqda yığılma bacarığı da artmış olur.
- İdmançıların (atletlərin) ürəyi. İntensiv idman və fiziki fəaliyyət nəticəsində sol mədəciyin divar qalınlığı, mədəcik həcmi və kütləsinin fizioloji böyüməsi baş verir ki, buna da "idmançı və ya atlet ürəyi" deyilir. İntensiv idman həm də müxtəlif növ və əsasən xoşxassəli aritmiyalarla da müşaiyət olunur. Lakin, əzəldən (alt xəstəlik) kardiomiopatiyası olan və intensiv idmanla məşğul olan idmançılarda fatal yəni ölümlə nəticələnə bilən aritmiyaların baş verməsi riski artmış olur. İdmançıların EKQ-lərində müəyyən edilən pozuntuların idmanla bağlı olmasını təsdiq etmək üçün kardiomiopatiyanın mümkünlüyü istisna edilməlidir. Qeyd edilməlidir ki, idmançılarda sol mədəciyin fizioloji hipertrofiyası HKM-dan fərqlidir. Belə ki, atletlərdə sol mədəciyin hipertrofiyası (LVH) adətən simmetrik və divar qalınlığı <12 mm-dən az olur (düzdür bəzi idmançılarda LVH 14-16 mm-dək ola bilər). Lakin, idmançıların müayinəsində yalnız LV divar qalınlığına əsaslanmaq yetərli deyil. Ürək-damar xəstəliyinə şübhəsi olan atletlərin müayinəsi mütləq detallı anamnezin toplanmasından, 12 aparmada EKQ-nin və EXOKQ əldə edilməsindən ibarət olmalıdır.
- Hipertrofiyanın digər səbəbləri. Ürək hipertrofiyasının digər səbəblərinə aşağıdakılar aiddir: genetik sindromlar (məs., Noonan sindromu), metabolik xəstəliklər (məs., Fridrix ataksiyası, Pomp xəstəliyi, AMP-kinaza), mitoxondriyal xəstəliklər, Fabri xəstəliyi, X xromosomla keçən resessiv qlikolipik depoziti xəstəliyi.
- HKM olmayan hallar. Yuxarıda qeyd olunduğu kimi mədəciklərin disfunksiyası ilə müşaiyət olunan miokard hipertrofiyası müxtəlif ürək-damar xəstəlikləri səbəbindən də inkişaf edə bilər. Lakin, ürək-damar xəstəlikləri səbəbindən ikincili inkişaf etmiş hipertrofiya 2006 və 2008-ci illərdə Amerika Ürək Assosiasiyası və Avropa Kardioloqlar Cəmiyyətinin kardiomiopatiya ilə bağlı tərifinə daxil deyil.
- Qeyri-infiltrativ xəstəliklər (birincili RKMP): idiopatik RKMP, irsi KMP, HKP, skleroderma, psevdoksantoma elasticum, diabetik kardiomiopatiya.
- İnfiltrativ xəstəliklər: amiloidoz, sarkoidoz, Quşer xəstəliyi, Hurler xəstəliyi, piy infiltrasiyası (piylənmə).
- İkincili RKMP əsasən hipertoniya xəstəliyi və ya ürəyin işemik xəstəliyinin ən son mərhələlərində inkişaf edə bilər.
- Toplanma xəstəlikləri: hemoxromatoz, Fabri xəstəliyi və qlikogen toplanması xəstəliyi.
- Digər xəstəliklər: endomiokardial fibroz, şüalanma, kimya terapiyası, hipereozinofiliya sindromu, karsinoid ürək xəstəliyi, metastazlarla müşaiyət olunan xərçəng, fibroz endokarditə səbəb olan dərmanlar (serotonin, metilsergid, erqotamin, civə, busulfan).
- Endokardial fibroelastoz. Endokardial fibroelastoz endomiokardial fibrozdan fərqləndirilməlidir. Endokardial fibroelastoz (EFE) sol mədəciyin endokardının diffuz qalınlaşması ilə xarakterizə olunur və fibroz və elastik toxumanın artımı (yayılması) səbəbindən baş verir. EFE iki forması vardır: dilatasiya ilə müşahidə edilən forma (Dilatasion KMP fenotipi; sol mədəcik böyümüş olur) və yığılmış forma (Restriktiv KMP fenotipi; sol mədəciyin boşluğu kiçik olur). EFE əsasən 1 yaşadək olan körpələrdə inkişaf edir və digər anadangəlmə ürək qüsurları (məs., sol mədəciyin çıxarıcı traktının obstruktiv patologiyaları, hipoplastik sol mədəcik) ilə yanaşı müşaiyət olunur. Düşünülür ki, EFE ürəyin zədələnməsinə səbəb olan müxtəlif faktorların təsirinə qarşı qeyri-spesifik cavab reaksiyası kimi inkişaf edir. Anoksiya, endokardit, virus infeksiyaları, genetik faktorların da EFE-nin inkişafında rol oynadığı düşünülür. İrsi EFE isə sistem karnitin çatışmazlığı ilə əlaqələndirilir. EFE həm də neonatal lupus (ananın autoanticismlərinin təsiri ilə inkişaf edən anadangəlmə ürək blokadası) ilə də əlaqələndirilir. EFE-nin dəqiq diaqnostikası üçün endomiokardial biopsiyanın aparılması tələb olunur.
- Sağ mədəciyin aritmogen displaziyası (kardiomiopatiyası). Sağ mədəciyin aritmogen kardiomiopatyası genetik ürək əzələsi xəstəliyidir və mədəciklərin aritmiyası və miokardın spesifik patologiyası ilə əlaqədardır. Sağ mədəcik miokardının sərbəst divarı (çox vaxt həm də sol mədəciyin divarı) fibroz və/və ya fibroz-piy toxuması ilə əvəzlənmiş olur. Sağ mədəciyin funksiyası pozulmuş olur, regional akineziya və ya diskineziyalar, ağır hallarda isə sağ mədəciyin qlobal dilatasiyası və disfunksiyası müşahidə edilir.
- Qeyri-kompakt sol mədəcik və ya süngərə bənzər sol mədəcik. Sol mədəciyin qeyri-kompakt kardiomiopatiyası çox nadir anadangəlmə patologiyadır. Onun əsasını ana bətnində olarkən miokard lifləri arasında patoloji sahələrin olması təşkil edir. Bu patologiya ürək çatışmazlığı, tromboemboliya və böyüklərdə mədəciklərin aritmiyası ilə əlaqələndirilir.
- Stress səbəbindən inkişaf etmiş kardiomiopatiya. Stress mənşəli kardiomiopatiya və ya takotsubo kardiomiopatiyası son zamanlar tez-tez müşahidə edilən sindromdur və ürəyin sol mədəciyinin orta seqmentləri və/və ya zirvə seqmentinin müvəqqəti sistolik disfunksiyası ilə xarakterizə olunur. Sindrom əsasən kəskin fiziki və ya emosional stress nəticəsində inkişaf edir.
- Sirroz mənşəli kardiomiopatiya. Məlumdur ki, alkoqolizm səbəbindən sirrozu olan xəstələrdə alkoqol mənşəli kardiomiopatiya ürək xəstəliyinin səbəblərindən biridir. Lakin, araşdırmalar göstərir ki, sirrozun özü də bir başa olaraq miokardın disfunksiyasına səbəb olur. Sirroz mənşəli kardiomiopatiyanın alt səbəbləri və kliniki mənzərəsi dəqiq müəyyən edilməyib, və onun müalicəsinə dair tövsiyələrdə də qeyri-müəyyənlik vardır. Guman edilir ki, sirroz vəziyyəti hiperdinamik qan dövranı, sodiumun artmış reabsorbsiyası, qarındaxili və sistem damarların genişlənməsi ilə xarakterizə olunur və qeyri-müəyyən patofizioloji mexanizmlərlə ürəyin disfunksiyasına səbəb olur. Sirrozla əlaqəli kardiomiopatiya sirrozu olan xəstələrin 40-50%-də müşahidə edilir. Sirrozla əlaqəli kardiomiopatiya həm də biliar atreziyası olan uşaqlarda da müəyyən edilir.
BIBLIOQRAFIYA.
- Abelmann WH. Classification and natural history of primary myocardial disease. Prog Cardiovasc Dis 1984; 27:73.
- Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Br Heart J 1980; 44:672.
- Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93:841.
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113:1807.
- Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29:270.
- Arbustini E, Narula N, Dec GW, et al. The MOGE(S) classification for a phenotype-genotype nomenclature of cardiomyopathy: endorsed by the World Heart Federation. J Am Coll Cardiol 2013; 62:2046.
- Elliott PM. Classification of cardiomyopathies: evolution or revolution? J Am Coll Cardiol 2013; 62:2073.
- Arnold JM, Liu P, Demers C, et al. Canadian Cardiovascular Society consensus conference recommendations on heart failure 2006: diagnosis and management. Can J Cardiol 2006; 22:23.
- WRITING COMMITTEE MEMBERS, Yancy CW, Jessup M, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013; 128:e240.
- Heart Failure Society of America, Lindenfeld J, Albert NM, et al. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 2010; 16:e1.
- Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med 1994; 331:1564.
- Devereux RB, Roman MJ, Paranicas M, et al. A population-based assessment of left ventricular systolic dysfunction in middle-aged and older adults: the Strong Heart Study. Am Heart J 2001; 141:439.
- Felker GM, Thompson RE, Hare JM, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med 2000; 342:1077.
- Braunwald E, Seidman CE, Sigwart U. Contemporary evaluation and management of hypertrophic cardiomyopathy. Circulation 2002; 106:1312.
- Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 1995; 92:785.
- St John Sutton MG, Lie JT, Anderson KR, et al. Histopathological specificity of hypertrophic obstructive cardiomyopathy. Myocardial fibre disarray and myocardial fibrosis. Br Heart J 1980; 44:433.
- Maron BJ, Wolfson JK, Roberts WC. Relation between extent of cardiac muscle cell disorganization and left ventricular wall thickness in hypertrophic cardiomyopathy. Am J Cardiol 1992; 70:785.
- Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107:2227.
- Rawlins J, Carre F, Kervio G, et al. Ethnic differences in physiological cardiac adaptation to intense physical exercise in highly trained female athletes. Circulation 2010; 121:1078.
- Corrado D, Pelliccia A, Bjørnstad HH, et al. Cardiovascular pre-participation screening of young competitive athletes for prevention of sudden death: proposal for a common European protocol. Consensus Statement of the Study Group of Sport Cardiology of the Working Group of Cardiac Rehabilitation and Exercise Physiology and the Working Group of Myocardial and Pericardial Diseases of the European Society of Cardiology. Eur Heart J 2005; 26:516.
- Thompson PD, Franklin BA, Balady GJ, et al. Exercise and acute cardiovascular events placing the risks into perspective: a scientific statement from the American Heart Association Council on Nutrition, Physical Activity, and Metabolism and the Council on Clinical Cardiology. Circulation 2007; 115:2358.
- Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med 1997; 336:267.
- Denfield, SW, Gajarski, RJ, Towbin, JA. Cardiomyopathies. In: Science and Practice of Pediatric Cardiology, 2nd Ed, Garson, A Jr, Bricker, JT, Fisher, DJ, Neish, SR (Eds), Williams and Wilkins, Baltimore 1998. p.1851.
- Lurie PR. Endocardial fibroelastosis is not a disease. Am J Cardiol 1988; 62:468.
- Tripp ME, Katcher ML, Peters HA, et al. Systemic carnitine deficiency presenting as familial endocardial fibroelastosis: a treatable cardiomyopathy. N Engl J Med 1981; 305:385.
- Nield LE, Silverman ED, Taylor GP, et al. Maternal anti-Ro and anti-La antibody-associated endocardial fibroelastosis. Circulation 2002; 105:843.
- Mahle WT, Weinberg PM, Rychik J. Can echocardiography predict the presence or absence of endocardial fibroelastosis in infants <1 year of age with left ventricular outflow obstruction? Am J Cardiol 1998; 82:122.
- Tworetzky W, del Nido PJ, Powell AJ, et al. Usefulness of magnetic resonance imaging of left ventricular endocardial fibroelastosis in infants after fetal intervention for aortic valve stenosis. Am J Cardiol 2005; 96:1568.
- Robinson JD, Del Nido PJ, Geggel RL, et al. Left ventricular diastolic heart failure in teenagers who underwent balloon aortic valvuloplasty in early infancy. Am J Cardiol 2010; 106:426.
- Ino T, Benson LN, Freedom RM, Rowe RD. Natural history and prognostic risk factors in endocardial fibroelastosis. Am J Cardiol 1988; 62:431.
- Basso C, Corrado D, Marcus FI, et al. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009; 373:1289.
- Milani A, Zaccaria R, Bombardieri G, et al. Cirrhotic cardiomyopathy. Dig Liver Dis 2007; 39:507.
- Møller S, Henriksen JH. Cirrhotic cardiomyopathy. J Hepatol 2010; 53:179.
- Zardi EM, Abbate A, Zardi DM, et al. Cirrhotic cardiomyopathy. J Am Coll Cardiol 2010; 56:539.
- Timoh T, Protano MA, Wagman G, et al. A perspective on cirrhotic cardiomyopathy. Transplant Proc 2011; 43:1649.
